摘要 基于第一性原理密度泛函理论探讨了四种典型SF6分解组分(SO2,SOF2,SO2F2以及H2S)分别在ZnO(0001)表面上的吸附以及传感性能。计算并比较了不同气体吸附所产生的吸附能、吸附距离、电荷转移、差分电荷密度,并通过能态密度计算探讨了气体分子与表面的化学相互作用。结果显示,四种气体分子均能与ZnO(0001)表面产生较强的化学吸附作用。除了SO2分子在吸附后还保留本身的分子结构外,其他三种分子均发生了不同程度的分解。SOF2与SO2F2表现为S-F键断裂而H2S表现为S-H键断裂。虽然计算结果显示ZnO(0001)表面对四种组分均有较强的吸附以及气敏性能,但因为较大的吸附能及气体的分解,ZnO对组分的恢复性能有待提升。本文能为基于ZnO及其改性材料的气敏传感器检测SF6分解组分提供理论基础及指导。
关键词:密度泛函理论 SF6分解组分 ZnO(0001)表面 气体吸附
六氟化硫(SF6)因其优秀的的绝缘性能,被广泛应用于各种大型电力设备中。然而,在设备生产或长期运行中,设备内部不可避免地存在各种绝缘缺陷,包括气体绝缘组合电器(Gas Insulated Swichgear, GIS)设备内的金属毛刺、自由导电微粒、绝缘子缺陷等。这些缺陷所造成的设备内部局部放电或局部过热现象会导致SF6气体的分解。在分解过程中,产生的一系列不稳定的低氟硫化物(SFx,x=1~5)会与设备内的微水微氧发生一系列的复杂化学反应,并生成一系列较稳定的化学组分,包括SO2、SOF2、SO2F2、H2S和HF等[1-2]。通过快速、低成本的方法检测这些分解产物的种类和含量,进而及时了解设备运行状态,保证电力系统稳定可靠运行十分重要[3-4]。
半导体型金属氧化物纳米材料拥有十分优秀的物理化学特性,包括高比表面积、高表面化学活性和优秀的光电特性。这些优异的性能使它们广泛应用于催化领域、储能领域、气敏与生物传感、光电器件等[5-6]。其中,过渡金属氧化物氧化锌因其具有合适的能隙宽度(约3.37eV)与结合能(60meV)、低成本、较高的化学稳定性、对特定气体有较高的化学活性,被广泛应用于各类型气敏材料中[7-8]。一般情况下,未经其他原子掺杂的氧化锌呈现n型半导体特性因而在合成过程中内部不可避免地出现氧空位。通过原子掺杂,或与其他化合物构成复合气敏材料,可显著提高对部分待测气体的气敏性能。对基于ZnO气敏传感器件,现阶段研究大多通过测量器件在不同气体环境中的电阻变化,来评估气敏传感器的性能。相关研究表明氧化锌对H2S气体有极低的检测极(达到1×10-9)[9-10],但大多数气敏机理的分析仅局限在定性阶段,且现阶段关于ZnO对于SF6的分解组分吸附及气敏机理分析较少。
基于第一性原理的密度泛函理论,被广泛应用于材料的气敏性能研究。通过该方法,就SF6分解组分的气敏性能,对包括金属氧化物(SnO2、TiO2等)[11-12]和碳材料(碳纳米管、石墨烯等)[13-14]等新型二维材料,已有研究给出较为严谨及系统的解释及分析,并且与实验有较高的契合度。所以,有必要采用基于第一性原理的量子化学方法详细探讨SF6分解组分在氧化锌表面的吸附行为及气敏性能。六方纤锌矿结构为ZnO稳定的化学结构[15]。每个Zn原子与相邻四个O原子进行成键与配位。Zn与O的化合价分别为+2与−2价。已有研究证明ZnO主要存在(![]() )、(
)、(![]() )、(
)、(![]() )、(0001)、(
)、(0001)、(![]() )等表面[16],并且通过理论计算,验证了五种ZnO表面稳定性(
)等表面[16],并且通过理论计算,验证了五种ZnO表面稳定性(![]() )>(
)>(![]() )>(
)>(![]() )/(0001)>(11
)/(0001)>(11![]() )[17]。虽然(
)[17]。虽然(![]() )为ZnO最稳定表面,但相关研究表明,ZnO生长方向倾向于c轴方向,使得(0001)等极化表面更容易暴露;(0001)等极化表面呈现出更优异的气敏性能[18]。因此本文选取ZnO(0001)面作为气体吸附表面,首先探讨了SO2、SOF2、SO2F2、H2S四种SF6分解组分分别在ZnO表面的较为稳定的多种吸附结构。之后针对每种吸附结构,探讨气体分子与表面的化学相互作用及体系的电子结构,计算差分电荷密度图(Charge Density Difference, CDD)、局域电子函数(Electron Localization Function, ELF)和电子能态密度(Density of States, DOS),详细分析吸附分子内原子与表面成键情况。本文从理论计算方面深入探讨ZnO表面与SF6分解组分气体分子相互作用行为,为开发基于ZnO及其复合材料的SF6分解组分化学传感器提供理论基础。
)为ZnO最稳定表面,但相关研究表明,ZnO生长方向倾向于c轴方向,使得(0001)等极化表面更容易暴露;(0001)等极化表面呈现出更优异的气敏性能[18]。因此本文选取ZnO(0001)面作为气体吸附表面,首先探讨了SO2、SOF2、SO2F2、H2S四种SF6分解组分分别在ZnO表面的较为稳定的多种吸附结构。之后针对每种吸附结构,探讨气体分子与表面的化学相互作用及体系的电子结构,计算差分电荷密度图(Charge Density Difference, CDD)、局域电子函数(Electron Localization Function, ELF)和电子能态密度(Density of States, DOS),详细分析吸附分子内原子与表面成键情况。本文从理论计算方面深入探讨ZnO表面与SF6分解组分气体分子相互作用行为,为开发基于ZnO及其复合材料的SF6分解组分化学传感器提供理论基础。
本文所有微观模型均在Materials Studio软件下构建,所有计算均在Dmol3模块下进行[19]。为了处理电子间的交换关联泛函,本文采用广义梯度近似(Generalized Gradient Approximation, GGA)方法中的Perdew-Burke-Ernzerhof(PBE)泛函进行近似[20]。采用P极化双数值轨道基组(Double Numerical Polarization, DNP)作为原子轨道线性组合方法(Linear Combination of Atomic Orbitals, LCAO)的基组函数。因Zn为过渡金属,采用包含相对论效应的模守恒赝势处理(DFT semi-core pseudopotential),轨道截断半径设置为4.5Å(1Å=1×10-10m)。鉴于吸附的分子与表面崔仔范德华弱相互作用力,所有模型均采用DFT-D2方法进行范德华力修正[21]。对于所有结构的几何优化弛豫,收敛标准设定为:两次几何优化能量差值小于1.0×10−5Ha(1Ha= 27.21eV),每个原子的受力小于0.002Ha/Å,每个原子的最大位移距离小于0.005Å。对于每步几何优化中的电子步迭代,两次计算能量差小于1.0×10−6Ha。
已有研究表明六方纤锌矿结构为ZnO的晶胞,参数为a=3.24Å,c=5.21Å,键长为1.98Å[15]。根据该晶胞结构,构建xy方向3×3超晶胞的四层ZnO(0001)表面,在表面几何优化以及吸附结构弛豫过程中,底部两层原子坐标位置完全固定,仅对顶部两层原子进行优化。经几何优化后ZnO(0001)表面结构如图1所示。其中,最顶部包括两种原子,四配位O原子(O4c)及三配位Zn原子(Zn3c)。对于所有关于ZnO(0001)表面的几何优化计算中,
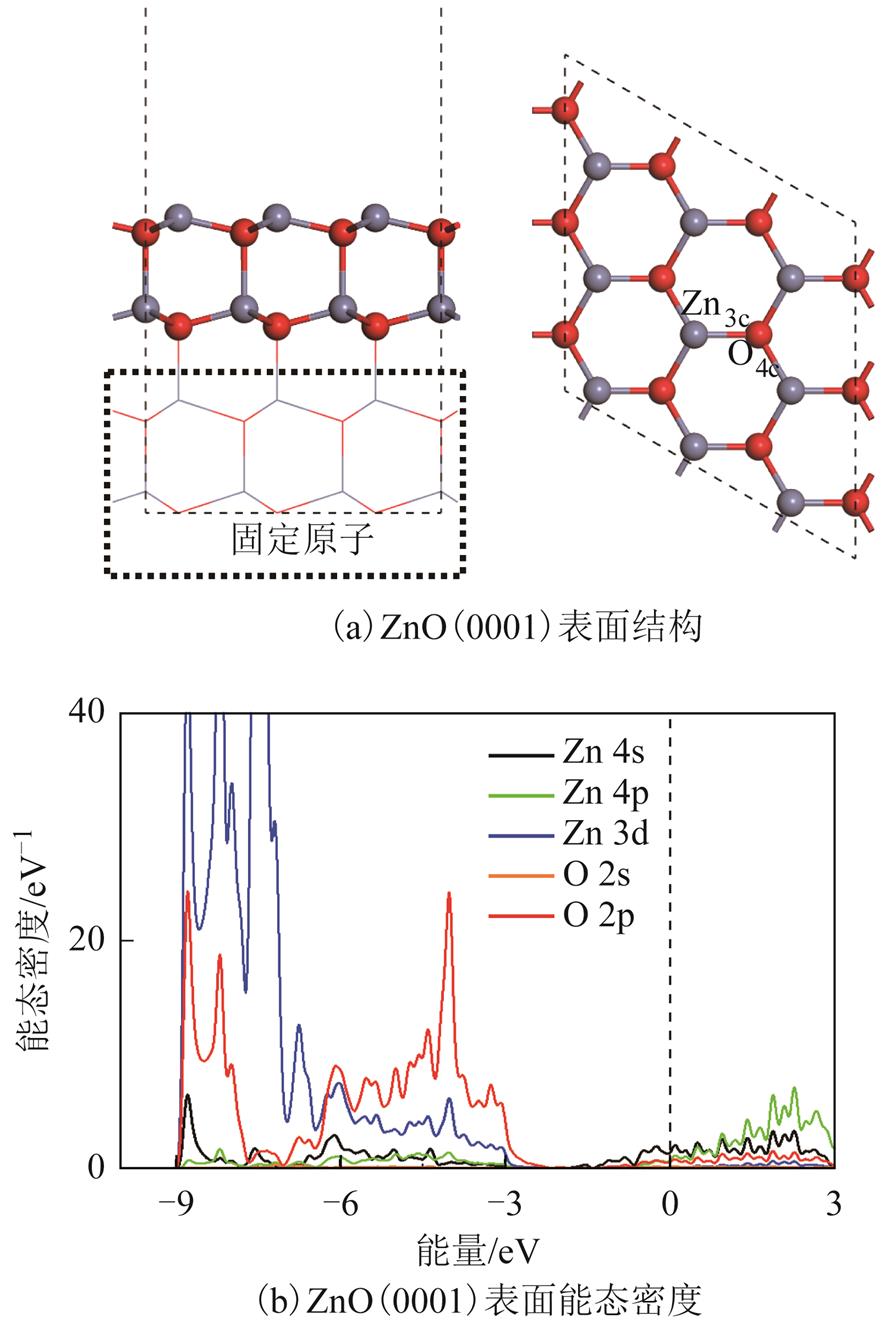
图1 ZnO(0001)表面结构与能态密度
Fig.1 Structure and DOS of ZnO(0001) surface
采用4×4×1的k点,对于几何优化后电子态密度的计算采用一个更精确的6×6×1的k点[22]。对于ZnO(0001)表面气体分子初始的吸附位点,本文设置为Zn顶部位点(TZn)以及O顶部位点(TO)。气体分子采用不同方向接近表面。最终对于单个气体分子吸附,获得多个局域总能量极小值的吸附结构。其中,单个气体分子在ZnO(0001)表面的吸附能计算式为
式中,![]() 为ZnO表面吸附单个气体分子后体系的总能量;
为ZnO表面吸附单个气体分子后体系的总能量;![]() 为吸附气体分子前ZnO表面的能量;
为吸附气体分子前ZnO表面的能量;![]() 为吸附前单个气体分子的能量。为分析气体分子与ZnO表面的化学相互作用,对体系的电荷密度、局域电子函数(Electron Localization Function, EL)和电子能态密度(Electronic Density of States, DOS)进行了计算与分析。
为吸附前单个气体分子的能量。为分析气体分子与ZnO表面的化学相互作用,对体系的电荷密度、局域电子函数(Electron Localization Function, EL)和电子能态密度(Electronic Density of States, DOS)进行了计算与分析。
在计算体系电荷密度后,得到三维差分电荷密度(Charge Density Difference, CDD)的计算公式为
式中,![]() 为ZnO表面吸附单个气体分子后体系的电荷密度;
为ZnO表面吸附单个气体分子后体系的电荷密度;![]() 为吸附气体分子前ZnO表面的电荷密度;
为吸附气体分子前ZnO表面的电荷密度;![]() 为吸附前单个气体分子的电荷密度。通过计算体系电荷密度,气体分子与表面间的电荷转移可通过Hirshfeld方法进行计算,计算式为[23]
为吸附前单个气体分子的电荷密度。通过计算体系电荷密度,气体分子与表面间的电荷转移可通过Hirshfeld方法进行计算,计算式为[23]
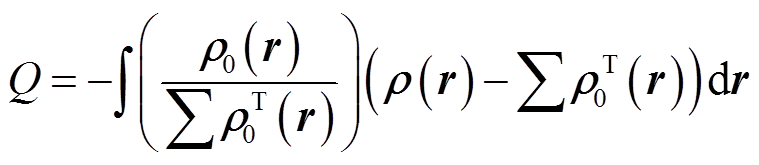 (3)
(3)
式中,![]() 为单个原子独立时的电荷密度;
为单个原子独立时的电荷密度;![]() 为整个计算体系的电荷密度。若结果为负数,则表示ZnO(0001)表面在吸附过程中失去电子,反之亦然。对于电子能态密度,本文进行了体系总的能态密度以及每个原子轨道的分波态密度计算。
为整个计算体系的电荷密度。若结果为负数,则表示ZnO(0001)表面在吸附过程中失去电子,反之亦然。对于电子能态密度,本文进行了体系总的能态密度以及每个原子轨道的分波态密度计算。
经过充分的几何优化后,ZnO(0001)表面结构如图1a所示。ZnO表面有两种配位原子,四配位O原子(O4c)与三配位(Zn3c)原子。O为满配位状态,而表面Zn包含悬空键,拥有较为活泼的化学性质。仅选取第一层表面原子作能态密度图。根据如图1b所示的能态密度图,在-9eV~-3eV范围内主要为Zn4s、Zn3d轨道与O2p轨道进行成键,组成ZnO表面的成键轨道,而大于0eV主要由Zn4p与Zn4s轨道组成ZnO的反键轨道。根据Hirshfeld电荷计算结果,表面Zn3c原子所带电荷为+0.31e,而表面O4c所带电荷为-0.33e。在Zn与O分别为单质时,Zn的价电子排布式为3d104s2,3d轨道与4s轨道均为全充满状态,而根据能态密度图,Zn的4s轨道存在空缺,说明电荷转移主要由Zn的4s轨道向O的2p轨道进行转移。几何优化后,表面ZnO的键长为1.92Å,小于体相ZnO键长(1.98Å),因此在形成(0001)面过程中,表面原子间键长缩短从而减小体系能量。
通过几何优化,获取了四种典型的SF6分解组分气体分子微观结构模型,结构及参数如图2所示,结果与现有报道[24-25]基本吻合,本文不再做详细讨论。
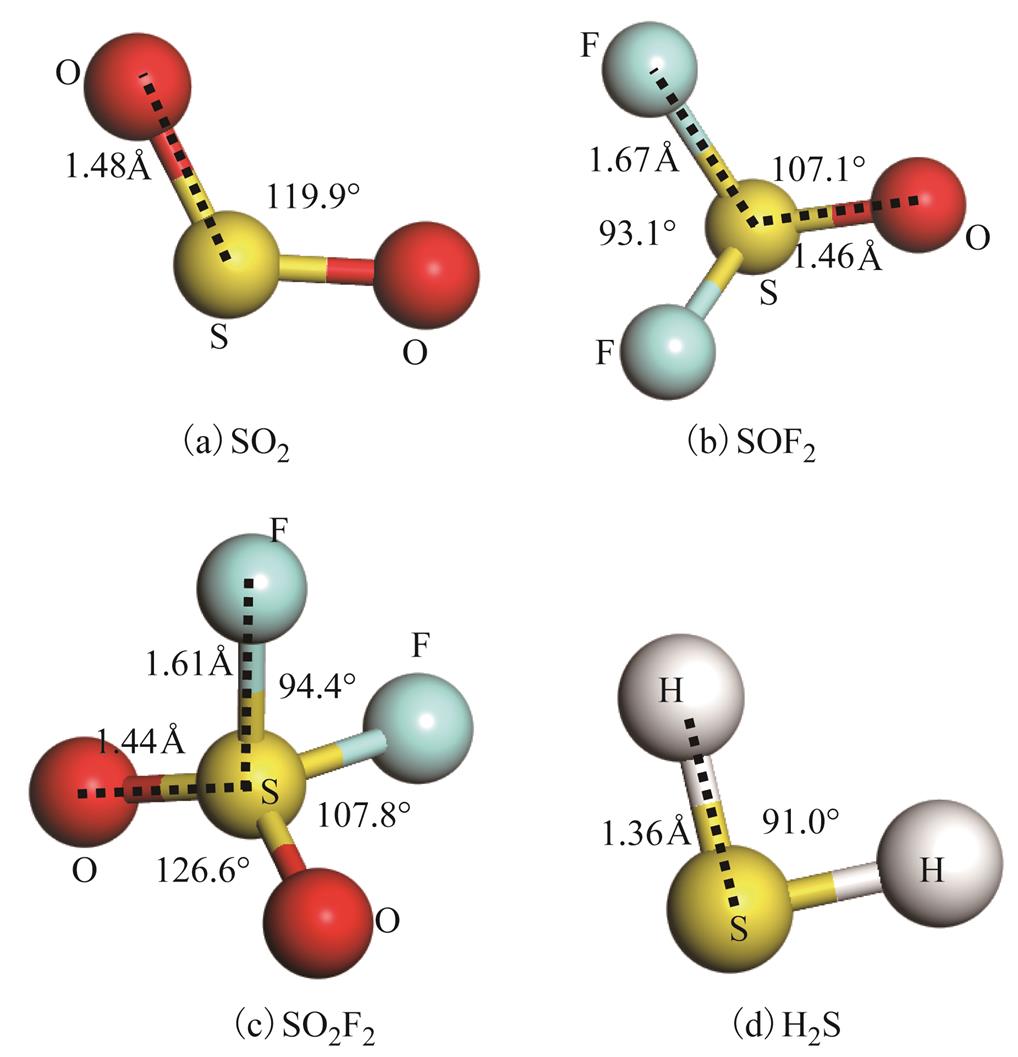
图2 四种典型SF6分解组分的几何结构
Fig.2 Structures of four types of typical SF6 decomposed products
根据第1节提及设置多种初始吸附结构,进行充分几何弛豫后,得到每种气体分别吸附在ZnO(0001)的能量极小值结构,每种气体均得到了两种较稳定的吸附结构。其中,对于SO2气体,得到的两种吸附结构如图3所示,其中,图3a结构显示,SO2分子几乎平行吸附在ZnO(0001)表面上,两个O原子倾向于接近表面Zn原子,另一个S原子同样距离Zn原子较近。所得到的吸附距离为1.98Å(O-Zn3c)与2.40Å(S-Zn3c)。对比1.98Å(O-Zn3c)的吸附距离与体相ZnO的键长(1.98Å)发现,两者几乎没有任何差异。因此,表面Zn3c与SO2中的O原子形成较明显化学键,并且该化学键强度几乎接近于体相ZnO中Zn-O键强度。更详细的成键强弱在2.3节中展开讨论。在体相ZnS中,Zn-S键长约为2.35Å[26],而SO2吸附后,表面Zn3c与S原子距离(2.40Å)仅略长于体相ZnS键长(2.35Å),因此可推测在SO2吸附后,表面Zn3c与S原子同样可能存在较强的化学相互作用,更细致的化学相互作用分析在2.3节中将会进一步讨论。该吸附结构所对应的吸附能为-3.10eV,并且SO2在吸附过程中得到电子。对于图3b吸附结构,吸附距离与图3a中吸附距离相当,吸附能(-3.08eV)略小于图3a的结构,但并无明显差异,并且电荷转移几乎与图3a中结构相同。因此,可以认为SO2在ZnO(0001)表面较大吸附能与电荷转移主要来源于SO2中S原子与O原子分别与表面的Zn3c原子产生较强烈的化学作用,并且该化学作用可能形成新的化学键。
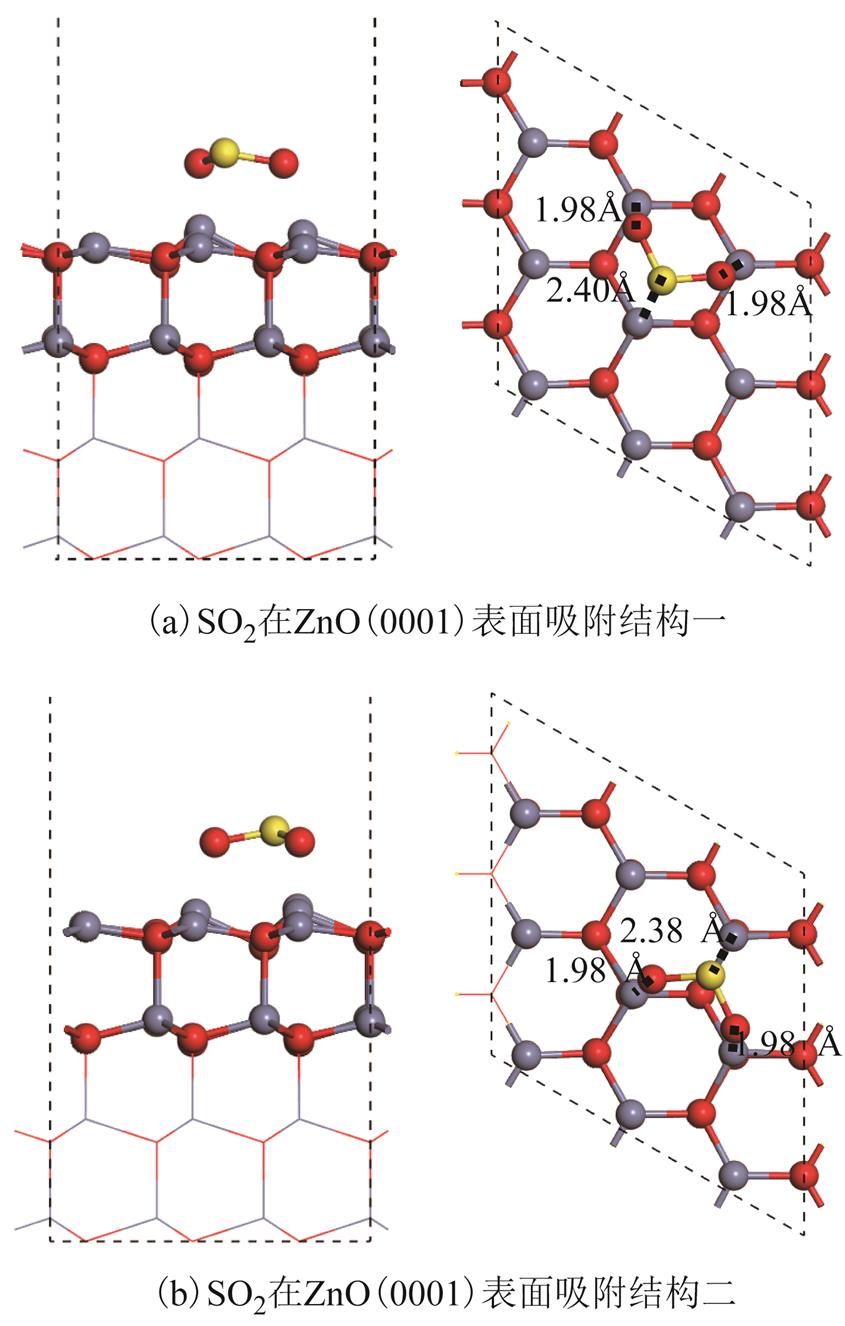
图3 SO2在ZnO(0001)表面吸附结构
Fig.3 Structures of SO2 adsorbed over ZnO(0001) surface
为更深入地探讨SO2与ZnO(0001)表面电荷转移,计算了差分电荷密度(CDD),如图4所示,图3与图4a、图4b结构分别对应。对比图4两种结构,电荷聚集区与电荷消散区均主要分布于SO2分子的周围,并且两者分布未有明显差异。电荷聚集区主要集中在SO2中的S原子与O原子周围,而消散区主要集中在SO2与ZnO(0001)表面之间,与分子内S-O键附近。对于分子内明显的电荷聚集与消散区,吸附过程明显改变了SO2分子内电荷分布。对于ZnO(0001)表面电荷分布,表面O4c原子下部有少量电荷聚集区,而Zn3c原子上部有明显的电荷消散区。因此在SO2吸附过程中,Zn3c原子会向O4c原子转移少量电子,而向SO2转移较多电子,从而导致了SO2的得电子行为。

图4 SO2在ZnO(0001)表面吸附差分电荷密度(等值面为0.01e/Å3)
Fig.4 Charge density difference (CDD) of SO2 adsorbed over ZnO(0001) surface (the value of isosurface is 0.01e/Å3)
对于SOF2在ZnO(0001)表面的吸附,本文获得了两种总能量极小值的吸附结构,如图5所示。图5a结构为SOF2分解成两个部分,其中一个为单个F原子,另一个为SOF片段。单个F原子吸附在三个Zn3c原子的中空位点中但离其中一个Zn3c原子较远。SOF片段吸附在另一个Zn3c原子组成的中空位点中,其中O原子、S原子与F原子分别与一个Zn3c原子靠近。其中,箭头为F原子在吸附过程中的移动方向。该结构带来-3.82eV与-0.48e电荷转移且ZnO表面失去电子。对于吸附距离而言,SOF片段中,Zn-S(2.48Å)、Zn-O(2.08Å)与Zn-F(2.27Å)的距离均略大于体相ZnO(1.98Å)、ZnS(2.35Å)与ZnF2(2.02Å/2.04Å)内距离[27]。因此,SOF中原子与表面Zn3c原子的相互作用可能相较于体相Zn化合物中的化学键偏弱。关于上述吸附原子与表面Zn的化学相互作用与原子轨道相互作用将在2.3节详细讨论。在图5b的吸附结构中,SOF2分子相比图5a结构发生更多的断键。两个S-F键均发生断裂,并且被分离出的两个F原子均吸附在Zn3c原子组成的中空位点中。SO片段也能稳定吸附于两个Zn3c原子上方,并且吸附距离相较于SOF片段更小,接近ZnO,ZnS体相化合物键长。图5b吸附结构的吸附能与电荷转移量明显大于图5a的结构。因此,认为F原子与表面Zn3c原子的化学相互作用所放出的能量,远大于一个S-F键断裂所吸收的能量。并且,该成键与断键的过程未表现出较明显的反应势垒,ZnO(0001)表面的SOF2分子中两个S-F键断裂的过程均属于自发反应。因SOF2分子在吸附前后发生明显的结构变形,所以未讨论差分电荷密度与分子内电荷分布变化。
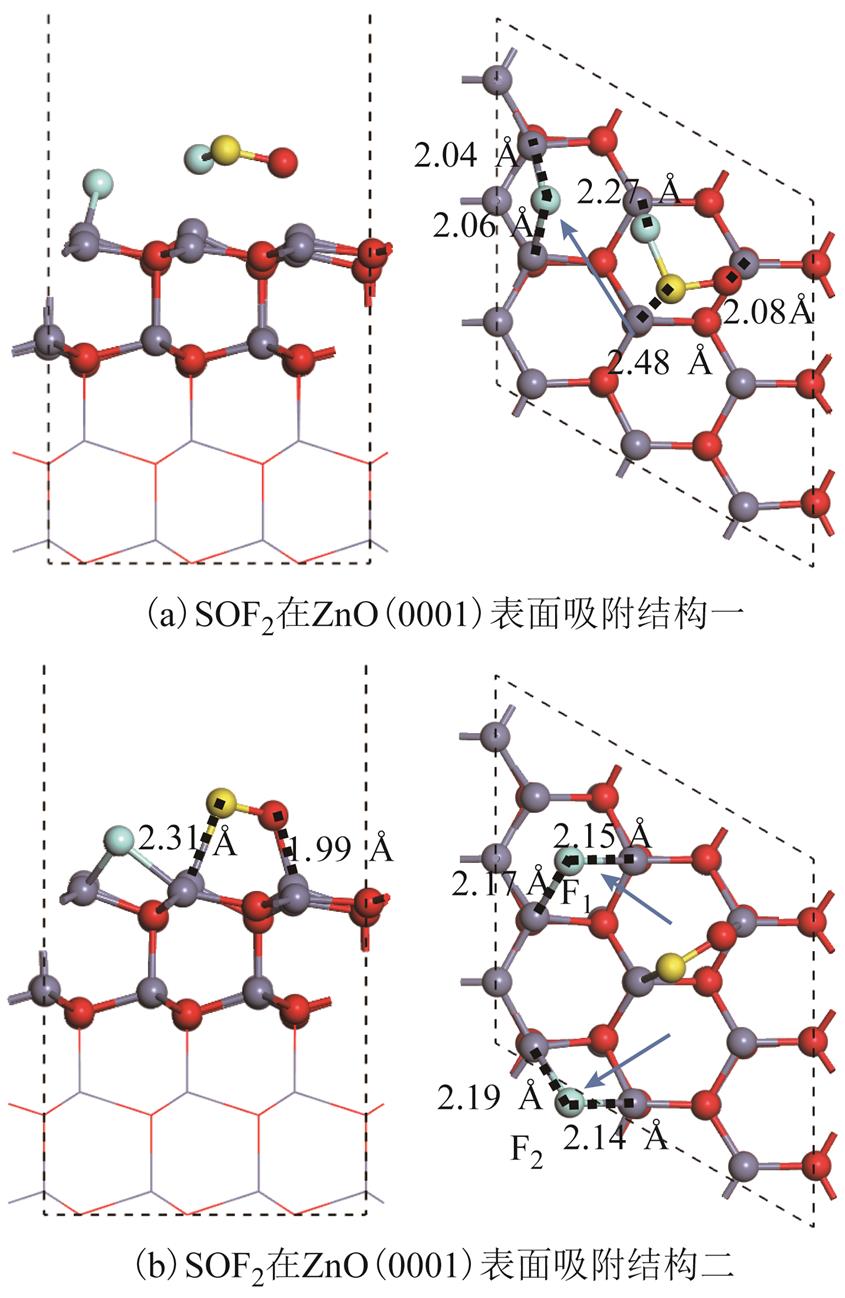
图5 SOF2在ZnO(0001)表面吸附结构
Fig.5 Structures of SOF2 adsorbed over ZnO(0001) surface
图6为经过几何优化后得到的两个SO2F2在ZnO(0001)表面的吸附结构。两种吸附结构均显示SO2F2分子发生了非常明显的分解并且与SOF2分解过程类似,无明显能量势垒,均属于自发进行。SO2F2分子中,两个S-F键均发生了断裂。与SOF2吸附结构相类似的是,分解出的F原子均位于Zn3c原子的中空位点中,而剩余的SO2片段几乎垂直吸附于ZnO(0001)表面,两个O原子靠近表面的Zn3c原子。此现象与单纯的SO2气体分子在表面的吸附略有不同。两种吸附结构均带来非常大的吸附能与电荷转移,但因为两种结构较类似,所以二者吸附能与电荷转移没有明显差别。因吸附结构相似,本文仅重点讨论图6a中的结构。对于SO2片段的吸附,O与Zn3c原子的距离(2.03Å/2.06Å)略大于ZnO体相键长(1.98Å),但S原子并未与周围Zn3c原子有较为明显的直接相互作用,因此,在周围F原子的影响下,SO2片段与ZnO(0001)表面的化学相互作用小于SO2单独吸附后与ZnO(0001)表面的相互作用。但通过SO2、SOF2的吸附过程对比,SO2F2的吸附会带来更大的吸附能与电荷转移,并且与ZnO(0001)表面有更强化学作用。
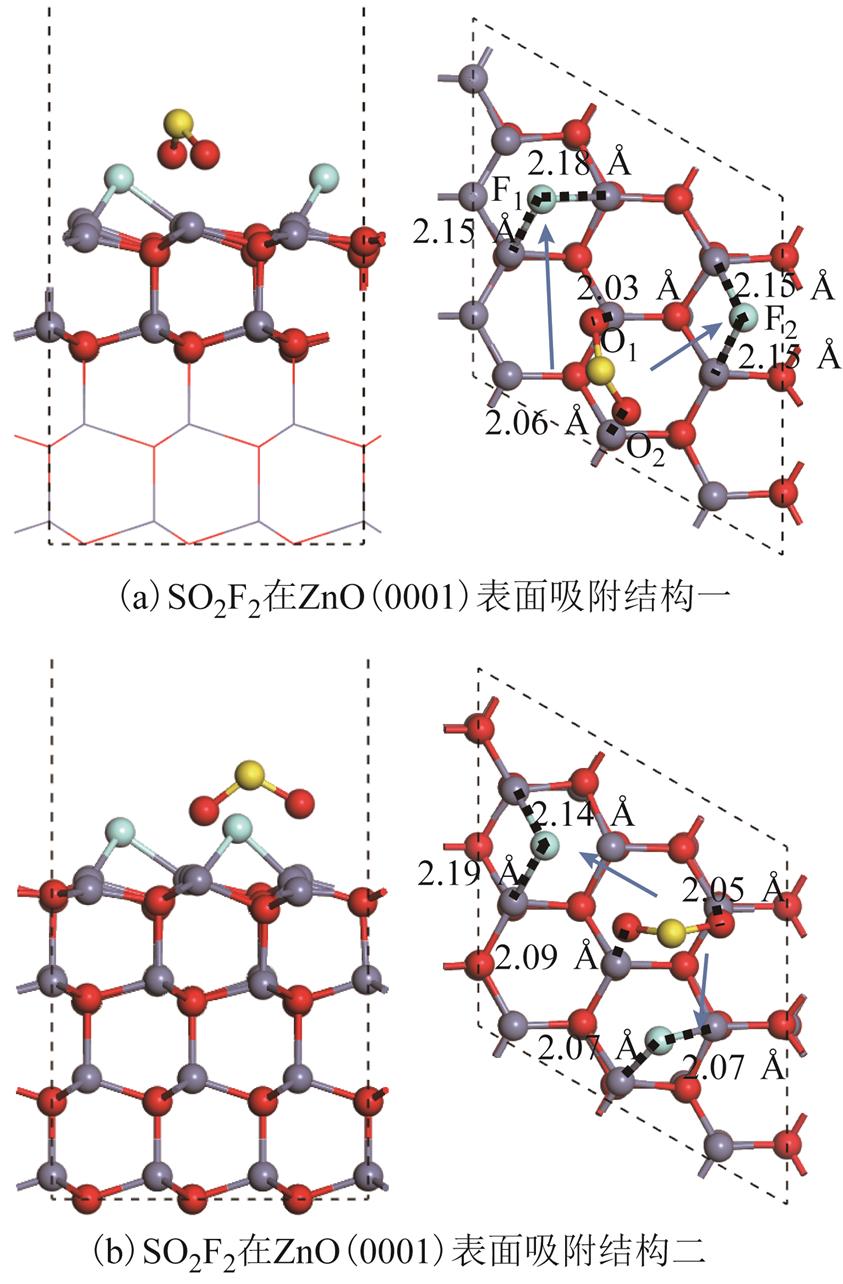
图6 SO2F2在ZnO(0001)表面吸附结构
Fig.6 Structures of SO2F2 adsorbed over ZnO(0001) surface
H2S分子在ZnO(0001)表面两个总能极小值的吸附结构如图7所示。对于图7a的结构,一个H-S键发生断裂,分离出的H原子吸附在某个Zn3c原子上方,另一个HS片段中S原子位于两个Zn3c原子上方中间。Zn3c与S原子的吸附距离(2.41Å/2.43Å)略微大于体相ZnS键长(2.35Å),其进一步化学作用将在2.3节探讨。此吸附结构已有相关学者进行了报道[28]。在本文中,此吸附结构带来-2.43eV吸附能以及-0.29e的电荷转移。在吸附过程中H2S分子发生断键并且得电子行为主要因为电子从Zn3c原子转移至单个的H原子以及HS片段中的S原子。不同于之前的研究,对于H2S的吸附结构,发现了一种吸附能更大的结构如图7b所示。该吸附结构相比图7a结构吸附能略微提升,至-2.46eV,电荷转移也略有提升。但整体结构仍然与图7a较为相似,一个H-S键发生断裂,H和S原子与表面Zn3c原子的成键情况也与图7a类似,区别在于HS片段在表面的方向存在差异。图7b结构中,HS片段中H原子倾向于在Zn3c原子的中空位点中,在吸附过程中HS片段也旋转了一定角度,如图7b箭头所示。
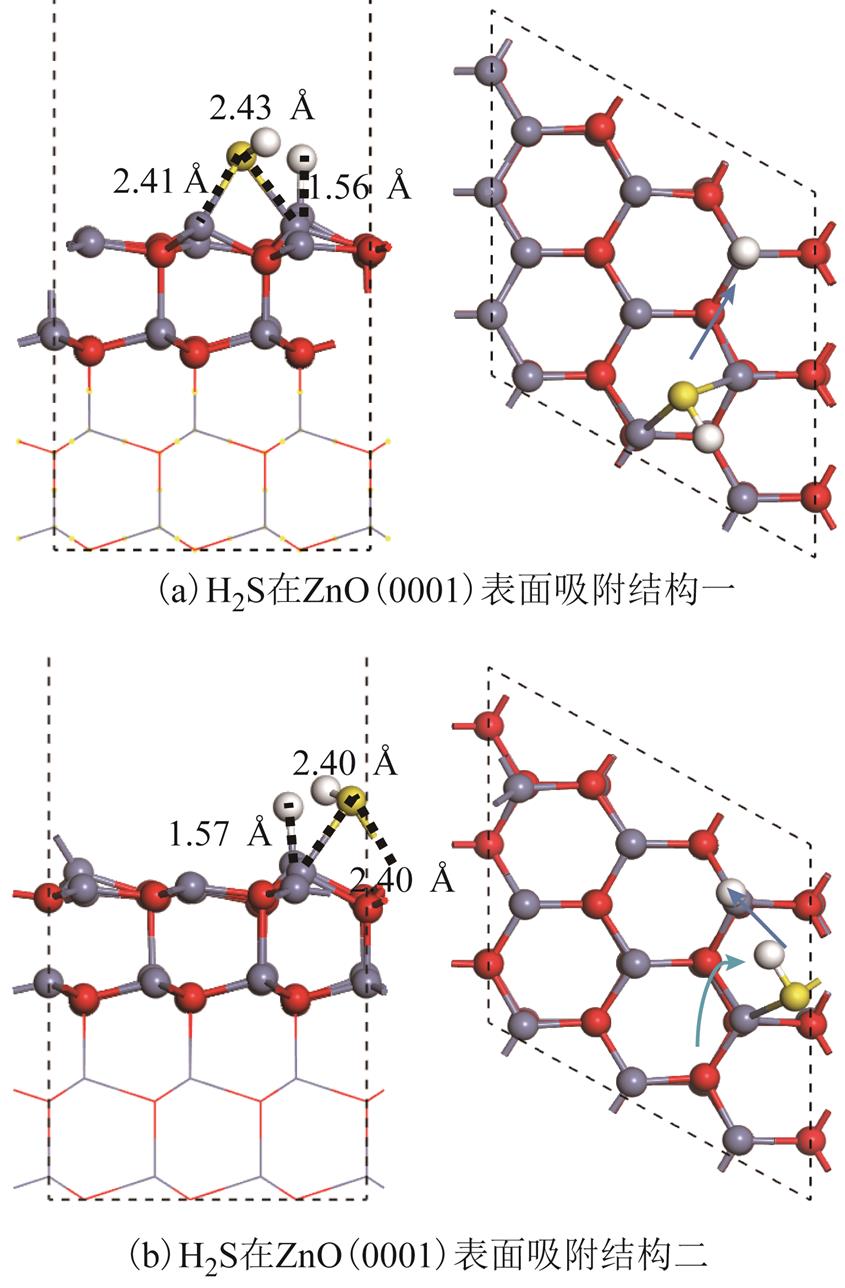
图7 H2S在ZnO(0001)表面吸附结构
Fig. 7 Structures of H2S adsorbed over ZnO(0001) surface
对于四种气体所计算出的八种总能极小值的吸附结构,吸附能与电荷转移汇总见表1。其中,SO2F2吸附有最大的吸附能以及较大的电荷转移,原因在于两个S-F键均发生断裂并且不存在能量势垒。SOF2有两种吸附情况,当一个S-F键断裂,吸附能与电荷转移均小于两个S-F键断裂的情况。对于SO2的吸附,虽然SO2分子结构未发生明显分解,但分子中S与O原子均与表面Zn3c原子有较强的成键趋势,因此放出能量。H2S的吸附虽然存在断键过程,但吸附能与电荷转移在四种气体里面最小。原因可能是HS的键能较小,并且H原子与表面Zn3c原子的相互作用比F、O或S与Zn3c原子作用较小,更多关于化学相互作用的分析在2.3节中展开。
表1 四种SF6分解组分在ZnO(0001)表面的吸附能与电荷转移对比
Tab.1 The adsorption energy and charge transfer of four types of SF6 decomposed products over ZnO(0001) surface
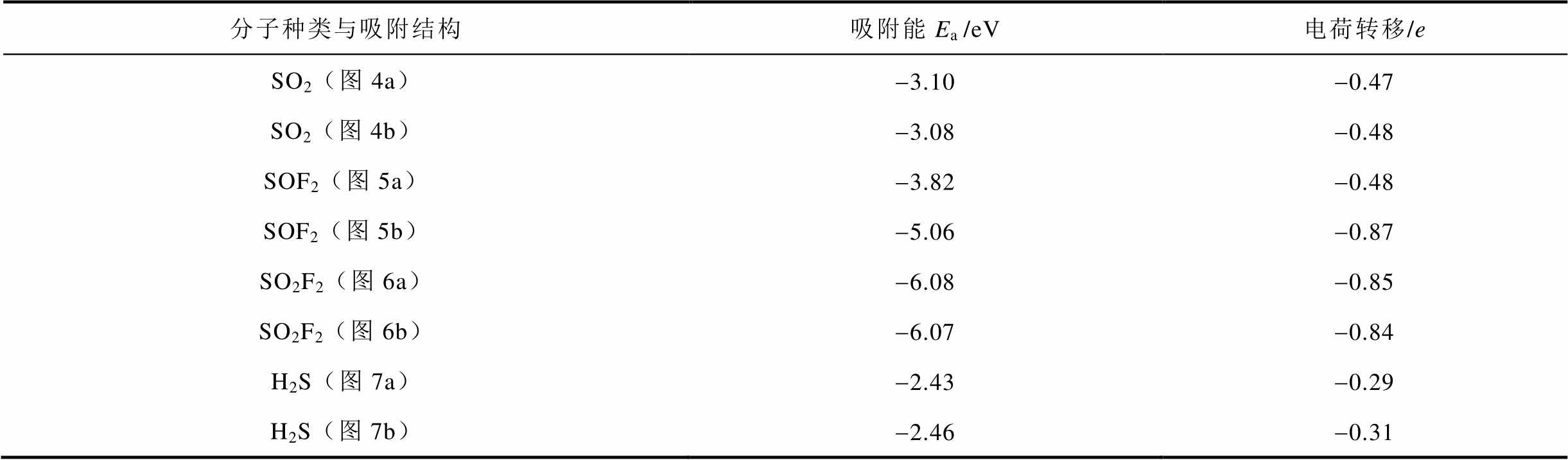
分子种类与吸附结构吸附能Ea /eV电荷转移/e SO2(图4a)-3.10-0.47 SO2(图4b)-3.08-0.48 SOF2(图5a)-3.82-0.48 SOF2(图5b)-5.06-0.87 SO2F2(图6a)-6.08-0.85 SO2F2(图6b)-6.07-0.84 H2S(图7a)-2.43-0.29 H2S(图7b)-2.46-0.31
为进一步探讨四种气体与ZnO(0001)表面的化学相互作用,本节通过计算能态密度(DOS)分析分子与原子轨道间的相互作用。对于SO2分子,本文对比了吸附前与吸附后的分子轨道,但因SOF2、SO2F2与H2S吸附后分子发生明显分解,对比分子轨道意义不大,所以本文仅探讨了分子内原子与表面原子的轨道相互作用。
对于SO2的吸附,本文仅详细对图3a中吸附结构进行DOS分析。SO2分子内总共有32个电子,构成了16个成键分子轨道。对于SO2详细的分子轨道分析已有较成熟的理论研究[29]。图8a给出了7个能量较高的成键分子轨道在DOS中的位置(6a1、4b2、7a1、2b1、5b2、1a2和8a1),与一个能量最低的反键轨道(3b1)。吸附前,8a1轨道作为SO2分子的最高占据态轨道(the Highest Occupied Molecular Orbital, HOMO)而3b1轨道作为最低未占据态轨道(the Lowest Unoccupied Molecular Orbital,LUMO)。吸附后,SO2分子轨道结构发生明显变化,变化主要在于4b2~8a1轨道能量范围发生明显交叠,并且反键轨道3b1部分进入费米能级以下。因此,对于SO2与ZnO(0001)表面的相互作用,主要包含了4b2~8a1轨道构型及能量分布发生明显改变,并且因为在该能量范围内呈现出多个新出现的峰,可能是由于在与表面相互作用过程中产生了多个新的轨道。反键轨道3b1部分进入费米能级之下,说明3b1轨道在吸附后存在占据态电子,也因此验证了SO2的得电子行为。分子内原子与表面原子轨道DOS如图8b所示。在-12eV附近,主要由S3s、O2s、O2p轨道与Zn3d轨道发生交叠作用,也是Zn3d与SO2的6a1分子轨道发生化学相互作用。在[-8eV, -5eV]区间内,轨道交叠主要存在于S3p、O2p与表面Zn3d轨道,也因为在该区域的轨道相互作用,使得SO2内电荷重新分配与分子轨道发生重组。在-1eV附近的峰主要为O2p、Zn4s与Zn4p的相互作用,也是SO2的反键轨道3b1与Zn4p的相互作用,并且成为整个体系的电子占据态。在整个体系的未占据态中,主要由Zn4s、Zn4p、S3p与O2p形成化学键,产生空轨道。综合来看,对于SO2内不同的原子轨道,在不同能量区间中,分别会与表面Zn的3d、4s、4p轨道分别产生化学相互作用,也使得SO2失去了原本分子轨道形貌。根据上述讨论,在[-12eV, -2eV]附近,主要为Zn的4s、3d轨道与吸附气体分子内S与O的原子轨道存在能态密度交叠,而在大于-2eV区域,主要为Zn的4p轨道与S、O的原子轨道有能态密度交叠。因此可得到不同能量范围内Zn原子轨道与气体分子均有较强的化学作用的结论。
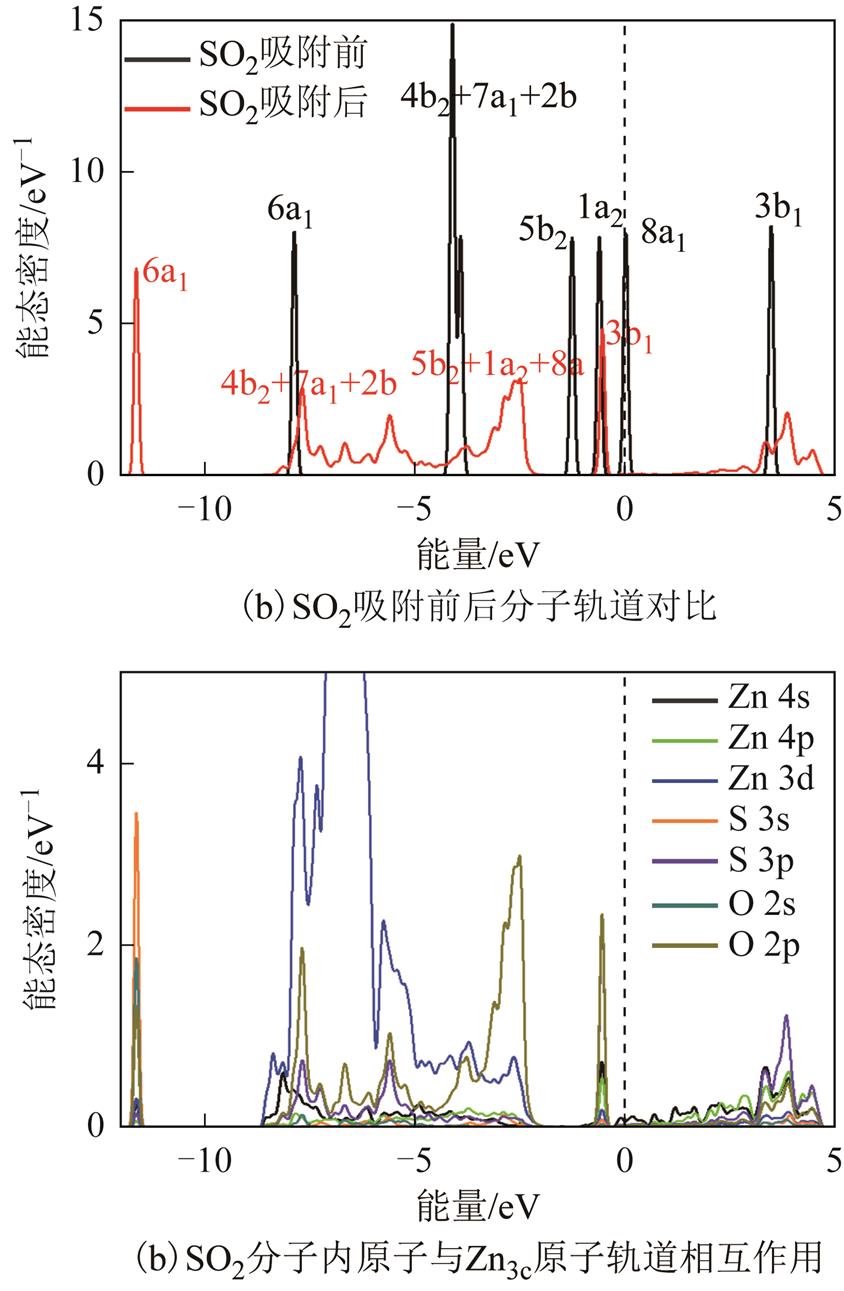
图8 SO2在ZnO(0001)表面吸附的DOS
Fig. 8 DOS of SO2 adsorbed over ZnO(0001) surface
SOF2有两种不同吸附结构,仅讨论吸附能更大的图5b结构的原子轨道化学相互作用。2.2小节中已经得到SOF2在吸附到表面后会发生分解,两个S-F键发生断裂,形成单独吸附的SO片段以及两个被吸附的F原子。因为分子发生明显分界,所以未比较吸附前后分子轨道的变化。吸附的SO片段的DOS如图9a所示。与SO2吸附类似的是,-12eV能量范围的峰主要为S3s、O2s、O2p与Zn3d轨道的相互作用产生。在[-8eV, -3eV]区间中,存在Zn4s、Zn3d、S3p与O2p能态交叠。与SO2吸附不同的是,SO片段的吸附在0eV附近,产生两个能态峰,两个峰均主要由S3p、O2p、Zn3d与Zn4p相互作用产生。未占据轨道主要由S3p、Zn4s与Zn4p组成,而O2p轨道所占比例较小,这也表现出了O原子在吸附过程中的得电子行为。为验证此行为,对O原子吸附前后进行Hirshfeld电荷分析,吸附前O原子所带电荷-0.21e而吸附后该值增加为-0.27e。对于分子分解产生单个吸附的F原子,选择其中一个F1原子进行分析,如图5b与图9b所示。其中,F2s原子轨道能量较低,在图9b能量范围内并未显示。在占据态[-7.5eV, -2eV]范围内,能态峰的交叠主要由Zn4s、Zn3d与F2p轨道产生,少量由Zn4p产生,而未占据态主要由Zn4s、Zn4p组成,F2p所占比例较小。本文继续进行原子Hirshfeld电荷计算,发现F1原子所带电荷为-0.29e,远大于吸附前SOF2分子内F原子电荷-0.13e,因此SOF2分解后表面Zn原子会向F2p轨道转移电子。另一个F2原子同样也有得电子行为,计算后带电量为-0.30e,与F1带电量相当。综上,吸附分子内原子轨道与表面Zn原子轨道存在明显的能态交叠,分子中S、O与F原子均与表面Zn原子有较强的化学相互作用。
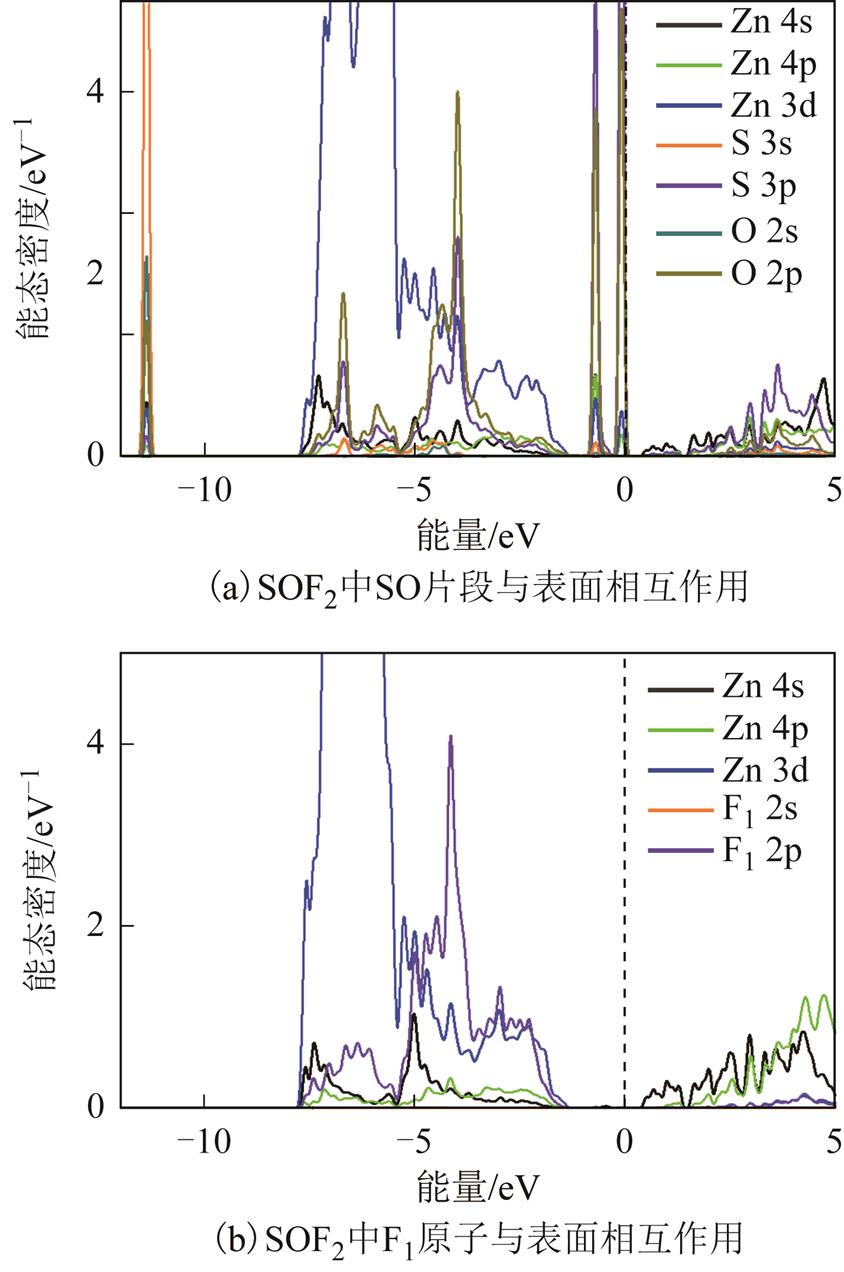
图9 SOF2在ZnO(0001)表面吸附的能态密度
Fig. 9 DOS of SOF2 adsorbed over ZnO(0001) surface
对于SO2F2的两种吸附结构,均呈现出分子分解后,两个F原子的单独吸附以及一个SO2片段的垂直吸附,因此仅选择一种结构(图6a)进行原子轨道讨论,如图10所示。对于SO2片段的垂直吸附,在-12eV左右的峰与表面单独吸附SO2分子产生的峰类似,而在-8eV附近存在一个新的峰,主要由Zn4s、Zn3d、S3p、O2s与O2p相互作用产生。在[-8eV,-5eV]范围内,S3p轨道有较多贡献,而在[-5eV, -3eV],S3p轨道贡献较少,O2p轨道贡献增加。对于0eV费米能级的峰,主要由S3p与O2p产生,而Zn贡献较少,因此该峰所对应的电子轨道主要分布在SO2片段上。对于非占据态轨道,主要由Zn4s、Zn4p、S3p与O2p组成,相比于单独SO2分子的吸附,SO2片段中O2p对于非占据态轨道贡献更大,对比O原子的Hirshfeld电荷,单独SO2分子吸附后,O原子呈现-0.32e负电荷,而SO2F2中SO2片段内O原子吸附后所带电荷量仅为-0.27e。因此SO2F2中分解出的SO2片段因为有周围F原子影响,得电子能力弱于SO2分子。而对于SO2F2分离出的F原子,与ZnO表面相互作用十分类似于SOF2中分离出的F原子,在此不再进行详细的分析。综上,分子内O原子与F原子均与表面Zn有直接化学相互作用,而S原子距离表面较远,并且与表面间有O原子间隔,只能通过与O原子轨道杂化后再与表面Zn发生化学相互作用。
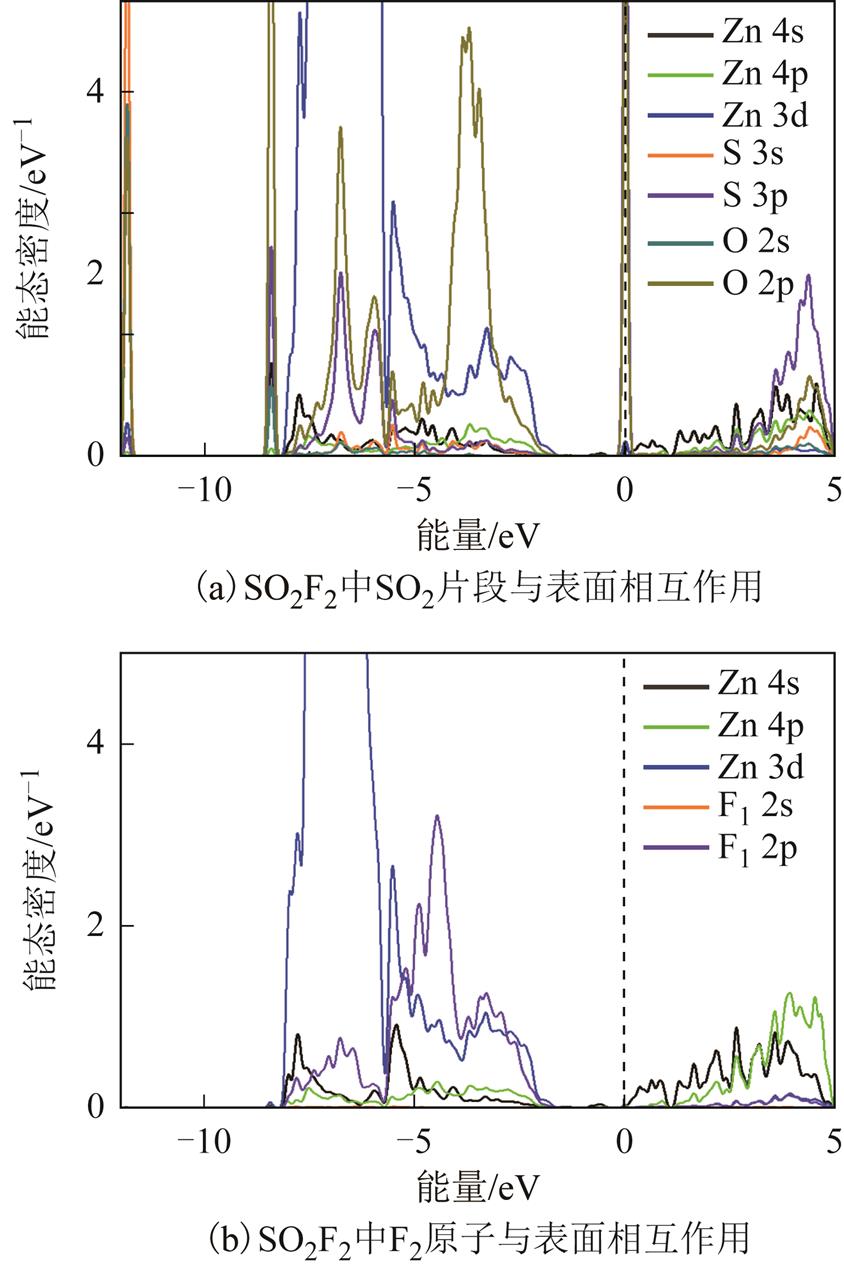
图10 SO2F2在ZnO(0001)表面吸附的能态密度
Fig. 10 DOS of SO2F2 adsorbed over ZnO(0001) surface
对于H2S在表面的吸附,选择吸附能及电荷转移量更大的图7b结构进行DOS分析,结果如图11所示。H2S吸附后分解为两个片段,H吸附在一个Zn3c原子正上方,而HS片段吸附在Zn3c之间的中空位点上。HS片段吸附后体系DOS如图11a所示。-14eV左右的峰由除了Zn4p轨道以外其他所选的五种原子轨道构成。在[-8eV, -5eV]能量范围内,Zn3d起主导作用,Zn4s、S3p与H1s相互之间也存在峰的重叠,该区域也主要由上述轨道形成相互的化学作用。在[-5eV, -3eV]区域,S3p起主导作用,并且H1s不参与该能量区域的成键。非占据态主要由Zn4s、4p和S3s构成,H1s轨道也占少量比例。因此表面Zn与S原子、Zn与分离出的H原子均存在较强的化学相互作用。对比吸附前后H2S分子内的电荷,吸附前H原子Hirshfeld电荷量为+0.05e,而吸附在Zn3c上方的H原子电荷量变为-0.17e,从Zn原子中得到大量电子。而S原子电荷量也从-0.11e增加至-0.16e,也印证了H2S吸附后从表面Zn原子的得电子行为。
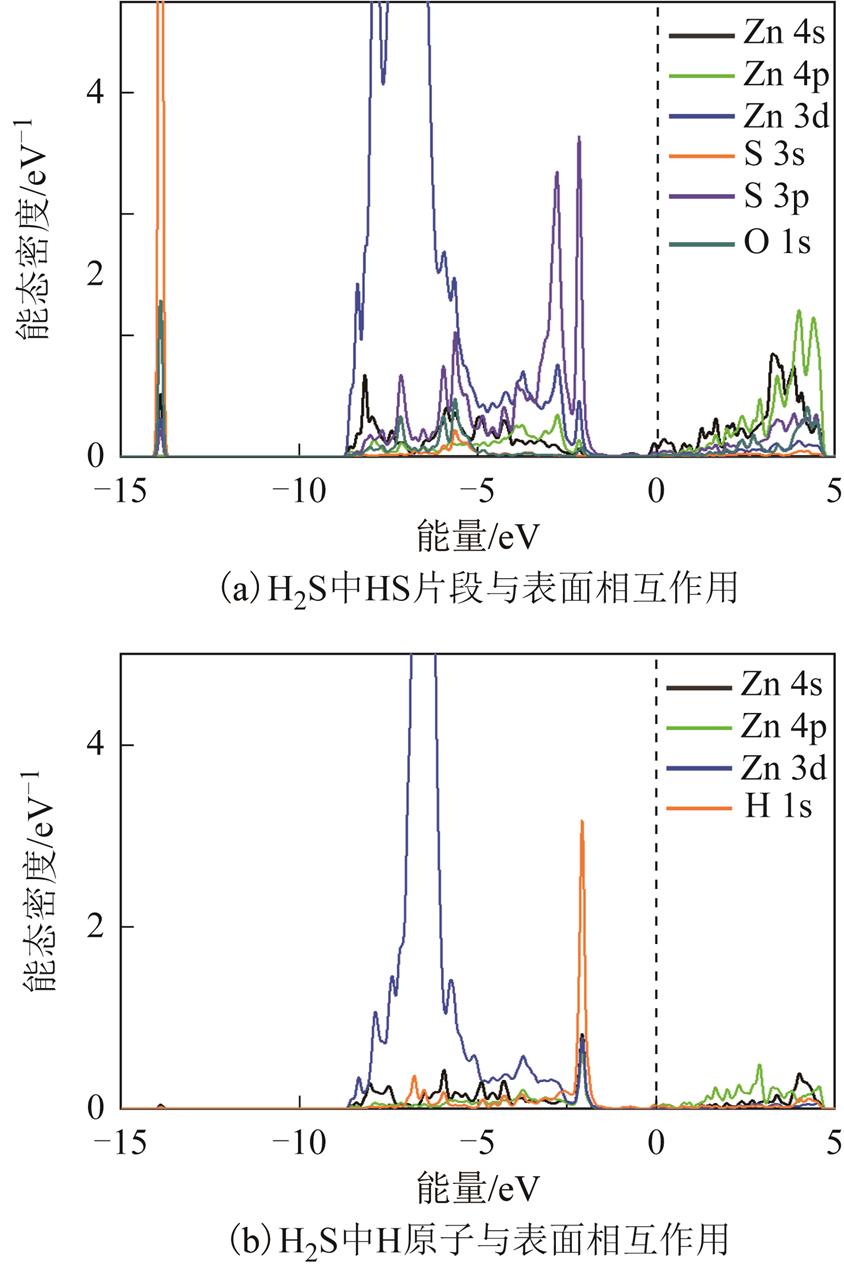
图11 H2S在ZnO(0001)表面吸附的能态密度
Fig. 11 DOS of H2S adsorbed over ZnO(0001) surface
考虑基于ZnO传感器检测SF6分解组分的实际应用,此节将对吸附能、电荷转移与气体传感器的灵敏度,选择性地进行对比讨论。ZnO主要为n型半导体材料[8],此特性表现在,当表面存在氧化性气体时,表面失去电子导致电阻率上升;当表面存在还原性气体时,表面得到电子导致电阻率下降。对于本文中涉及到的四种气体,SOF2电荷转移量最大,达到0.87e,SO2F2次之,为0.85e,SO2与H2S较小,为0.48e与0.31e。因此,在相同吸附量,并且不考虑氧气、水等其他物质的吸附与反应时,四种气体均会造成基于ZnO传感器的电阻率下降。考虑到SO2F2吸附能明显大于SOF2,在表面会更易分解并且吸附量更大,对SO2F2灵敏度将会最大,SOF2次之。相比之下对SO2与H2S灵敏度较小,SO2略大于H2S。综上,在(0001)表面大量暴露的ZnO基传感器中,不考虑杂质气体的影响,对于SO2F2灵敏度最高,H2S灵敏度最低。因SOF2、SO2F2与H2S气体存在表面分解行为,在无外界干扰时,传感器恢复特性并不理想,需要增加外界条件提升恢复性能,增加传感器寿命,如当传感器结束一个使用周期时,通过在富氧气环境中加高热或者加高功率紫外,加速被吸附气体的脱附,增加传感器可重复性,使得传感器具有更高的工程使用价值。
本文构建了ZnO(0001)表面并基于第一性原理密度泛函理论探讨了四种典型的SF6分解组分(SO2、SOF2、SO2F2与H2S)在表面的吸附结构,对比了不同气体分子吸附所产生的吸附能、电荷转移,并通过分析分子内与表面的原子轨道在一定能量区间产生的交叠,进一步探讨了吸附中的化学相互作用,进而对比ZnO对不同气体的气敏性能强弱。
1)四种气体在ZnO(0001)表面的吸附均能产生较大的吸附能与电荷转移,吸附能大小为SO2F2>SOF2>SO2>H2S,电荷转移大小为SO2F2≈SOF2>SO2>H2S。
2)对于四种气体的吸附,对比气体中原子与表面Zn的距离,并讨论原子轨道的能态密度交叠作用,四种吸附均与表面产生较强烈化学相互作用。
3)其中SOF2、SO2F2与H2S的吸附发生了明显的分解过程,SOF2与SO2F2呈现出S-F键的断裂,而H2S为S-H键断裂。分解后分散的原子与片段均能稳定吸附于ZnO(0001)表面上。
4)因为存在较大的吸附能以及较强的化学相互作用,虽然ZnO均可能对上述四种SF6分解组分产生较高的响应,但存在较困难的脱附行为,需要人为采取相应措施提升传感器的可重复利用性。
本文从理论上分析对比了ZnO(0001)表面对四种气体吸附及气敏性能,也指出了基于ZnO气敏材料检测SF6组分可能存在的缺陷,为后续实验研究以及气敏传感器性能的改进进行了展望并提出指导意见。
参考文献
[1] 唐炬, 杨东, 曾福平, 等. 基于分解组分分析的SF6设备绝缘故障诊断方法与技术的研究现状[J]. 电工技术学报, 2016, 31(20): 41-54. Tang Ju, Yang Dong, Zeng Fuping, et al. Research status of SF6 insulation equipment fault diagnosis method and technology based on decomposed components analysis[J]. Transactions of China Electrotechnical Society, 2016, 31(20): 41-54.
[2] 乔胜亚, 周文俊, 王勇, 等. 典型吸附剂对GIS固体绝缘介质放电特征气体变化规律影响[J]. 电工技术学报, 2018, 33(19): 4627-4635. Qiao Shengya, Zhou Wenjun, Wang Yong, et al. Effect of typical adsorbents on gas change characteristics of gas insulated switchgear solid insulation dielectric[J]. Transactions of China Electrotechnical Society, 2018, 33(19): 4627-4635.
[3] 唐念, 乔胜亚, 李丽, 等. HF和H2S作为气体绝缘组合电器绝缘缺陷诊断特征气体的有效性[J]. 电工技术学报, 2017, 32(19): 202-211. Tang Nian, Qiao Shengya, Li Li, et al. Validity of HF and H2S as target gases of insulation monitoring in gas insulated switchgear[J]. Transactions of China Electrotechnical Society, 2017, 32(19): 202-211.
[4] 任敬国, 辜超, 师伟, 等. 1100kV GIS设备主回路绝缘试验电气参量估算方法研究[J]. 电力系统保护与控制, 2018, 46(3): 103-109. Ren Jingguo, Gu Chao, Shi Wei, et al. Research on evaluation method of electric parameters of 1100 kV GIS main circuit insulation test [J]. Power System Protection and Control, 2018, 46(3):103-109.
[5] Wenderich K, Mul GMethods. Mechanism, and applications of photodeposition in photocatalysis: a review[J]. Chemical Reviews, 2016, 116(23): 14587-14619.
[6] Zhang Peng, Wu Jiang, Zhang Ting, et al. Perovskite solar cells with ZnO electron-transporting materials[J]. Advanced Materials, 2018, 30(3): 1703737.
[7] Dral A P, Johan E. 2D metal oxide nanoflakes for sensing applications: review and perspective[J]. Sensors and Actuators B: Chemical, 2018, DOI: 10.1016/j.snb.2018.05.157 .
[8] Zhu Ling,Zeng Wen.Room-temperature gas sensing of ZnO-based gas sensor: a review[J]. Sensors and Actuators A: Physical, 2017, DOI: 10.1016/j.snb.2017. 10.021 .
[9] Wang Caihong, Chu Xiangfeng, Wu Mingmei. Detection of H2S down to ppb levels at room temperature using sensors based on ZnO nanorods[J]. Sensors and Actuators B: Chemical, 2006, 113(1): 320-323.
[10] Hosseini Z S, Mortezaali A. Room temperature H2S gas sensor based on rather aligned ZnO nanorods with flower-like structures[J]. Sensors and Actuators B: Chemical, 2015, 207: 865-871.
[11] Wang Dawei, Wang Xiaohua, Yang Aijun, et al. A first principles theoretical study of the adsorption of SF6 decomposition gases on a cassiterite (110) surface[J]. Materials Chemistry and Physics, 2018, 212: 453-460.
[12] 张晓星, 董星辰, 陈秦川. 锐钛矿型 (101) 晶面吸附SF6局部放电分解组分的气敏机理分析[J]. 电工技术学报, 2017, 32(3): 200-209. Zhang Xiaoxing, Dong Xingchen, Chen Qinchuan. Gas sensing mechaism analysis of SF6 decomposed gases adsorption on anatase (101) surface under partial discharge[J]. Transactions of China Electrotechnical Society, 2017, 32(3): 200-209.
[13] 张晓星, 方佳妮, 崔豪, 等. 碳纳米管传感器吸附机理及对SF6分解组分检测应用综[J]. 中国电机工程学报, 2018, 38(16): 4926-4941. Zhang Xiaoxing, Fang Jiani, Cui Hao, et al. Review: adsorption principle and application of carbon nanotubes to SF6 decomposition components[J]. Proceedings of the CSEE, 2018, 38(16): 4926-4941.
[14] 张晓星, 黄蓉, 喻蕾, 等. 基于第一性原理的金掺杂石墨烯对SF6分解组分的气敏分析[J]. 中国电机工程学报, 2017, 37(6): 1828-1834. Zhang Xiaoxing, Huang Rong, Yu Lei, et al. Gas sensing analysis properties of Au-Graphene to SF6 decomposition products based on a first principle study[J]. Proceedings of the CSEE, 2017, 37(6): 1828-1834.
[15] Xu Y N, Ching W Y. Electronic, optical, and structural properties of some wurtzite crystals[J]. Physical Review B, 1993, 48(7): 4335-4351.
[16] Diebold U, Koplitz L V, Dulub O. Atomic-scale properties of low-index ZnO surfaces[J]. Applied Surface Science, 2004, 37(1-4): 336-342.
[17] Tang Qianlin, Luo Qinghong. Adsorption of CO2 at ZnO: a surface structure effect from DFT+U calculations[J]. The Journal of Physical Chemistry C, 2013, 117(44): 22954-22966.
[18] Saputro A G, Agusta M K, Yuliarto B, et al. Selectivity of CO and NO adsorption on ZnO (0002) surfaces: a DFT investigation[J]. Applied Surface Science, 2017, 410: 373-382.
[19] Delley B. From molecules to solids with the DMol3 approach[J]. The Journal of Chemical Physics, 2000, 113(18): 7756-7764.
[20] Perdew J P, Burke K, Ernzerhof M. Generalized gradient approximation made simple[J]. Physical Review Letters, 1996, 77(18): 3865.
[21] Grimme S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction[J]. Journal of Computational Chemistry, 2006, 27(15): 1787-1799.
[22] Monkhorst H J, Pack J D. Special points for Brillouin-zone integrations[J]. Physical Review B, 1976, 44(2): https://doi.org/10.1103/PhysRevB.16.1746.
[23] Hirshfeld F L. Bonded-atom fragments for describing molecular charge densities[J]. Theoretica Chimica Acta, 1977, 44(2): 129-138.
[24] Fu Yuwei, Yang Aijun, Wang Xiaohua, et al. Theoretical study of the neutral decomposition of SF6 in the presence of H2O and O2 in discharges in power equipment[J]. Physics D: Applied Physics, 2016, 49(38): 385203.
[25] Zhang Xiaoxing, Chen Dachang, Cui Hao, et al. Understanding of SF6 decompositions adsorbed on cobalt-doped SWCNT: a DFT study[J]. Applied Surface Science, 2017, 420: 371-382.
[26] Kisi E H, Elcombe M M. u parameters for the wurtzite structure of ZnS and ZnO using powder neutron diffraction[J].Acta Crystallographica Section C: Structural Chemistry, 1989, 45(12): 1867-1870.
[27] O'Toole N J, Streltsov V A. Synchrotron X-ray analysis of the electron density in COF2 and ZnF2[J]. Acta Crystallographica Section B: Structural Science, 2001, 57(2): 128-135.
[28] Yin Gaiyu, Ding Kaining, Li Junqian. The first-principles calculations of H2S adsorption and decomposition on the ZnO (0001) surface[J]. Chinese Journal of Structural Chemistry, 2010, 29(8): 1139-1146.
[29] Rothenberg S, Schaefer III H F. Theoretical study of SO2 molecular properties[J]. The Journal of Chemical Physics, 1970, 53(8): 3014-3019.
Density Functional Theory Study of SF6 Decomposed Products Over ZnO(0001) with Gas Sensing Properties
Abstract In this work, the first-principles with density functional theory was adopted to investigate the adsorption and gas sensing properties of ZnO(0001) surface toward four types of typical SF6 decomposed products (SO2, SOF2, SO2F2 and H2S). The adsorption energy, adsorption distance, charge transfer, charge density difference (CDD), electron localization function (ELF) were calculated and compared. And also, the chemical interactions between the adsorbed gas molecule and the surface were analyzed based on the density of states (DOS) results. The results show that all these four types of molecules bring strong chemisorption behavior. Only SO2 remains its original structure but other three types of molecules all break apart with different extent. The S-F bonds in SOF2 and SO2F2 and the S-H bond rupture during the adsorption and the bond rupture processes do not show obvious energy barrier and are spontaneous reactions. Although the results in this study show that the ZnO(0001) surface exhibits strong chemical interactions and gas sensing properties to these four types of SF6 decomposed products, the large adsorption energy and the decomposition of adsorbed gas molecule bring relatively weak recovery properties. This study can provide theoretical basis and guidance for ZnO and its modified material based gas sensor to detect SF6 decomposed products.
keywords:Density functional theory, SF6 decomposed products, ZnO(0001) surface, gas adsorption
DOI:10.19595/j.cnki.1000-6753.tces.190069
中图分类号:TM85
王邸博 男,1988年生,博士,工程师,研究方向为高压电器可靠性及其应用技术。E-mail:wangdb@csg.cn
陈达畅 男,1993年生,博士,研究方向为电气设备在线监测与故障诊断。E-mail:dachangchen@whu.edu.cn(通信作者)
国家自然科学基金资助项目(51777144)。
收稿日期2019-01-13
改稿日期 2019-06-04
(编辑 郭丽军)